Reactions of Alcohols
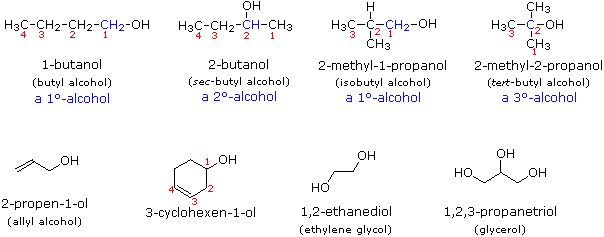
Alcohol Nomenclature
In the IUPAC system of nomenclature, functional groups are normally designated in one of two ways.
The presence of the function may be indicated by a characteristic suffix and a location number.
This is common for the carbon-carbon double and triple bonds which have the respective suffixes
ene and yne. Halogens, on the other hand, do not have a suffix and are named as
substituents, for example: (CH3)2C=CHCHClCH3 is
4-chloro-2-methyl-2-pentene. If you are uncertain about the IUPAC rules for nomenclature you
should review them now.
Alcohols are
usually named by the first procedure and are designated by an ol suffix, as in ethanol,
CH3CH2OH (note that a locator number is not needed on a two-carbon chain).
On longer chains the location of the hydroxyl group determines chain numbering. For example:
(CH3)2C=CHCH(OH)CH3 is 4-methyl-3-penten-2-ol. Other examples of
IUPAC nomenclature are shown below, together with the common names often used for some of the
simpler compounds. For the mono-functional alcohols, this common system consists of naming the
alkyl group followed by the word alcohol. Alcohols may also be classified as
primary, 1°, secondary, 2° & tertiary, 3°, in the same manner as alkyl
halides. This terminology refers to alkyl substitution of the carbon atom bearing the hydroxyl
group (colored blue in the illustration).
Many functional groups have a characteristic suffix designator, and only one such suffix (other than "ene" and "yne") may be used in a name. When the hydroxyl functional group is present together with a function of higher nomenclature priority, it must be cited and located by the prefix hydroxy and an appropriate number. For example, lactic acid has the IUPAC name 2-hydroxypropanoic acid.
Compounds incorporating a C–S–H functional group are named thiols or mercaptans. The IUPAC name of (CH3)3C–SH is 2-methyl-2-propanethiol, commonly called tert-butyl mercaptan. The chemistry of thiols will not be described here, other than to note that they are stronger acids and more powerful nucleophiles than alcohols.
Alcohols
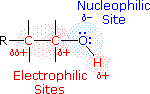
The functional group of the alcohols is the hydroxyl group, –OH. Unlike the alkyl halides, this group has two reactive covalent bonds, the C–O bond and the O–H bond. The electronegativity of oxygen is substantially greater than that of carbon and hydrogen. Consequently, the covalent bonds of this functional group are polarized so that oxygen is electron rich and both carbon and hydrogen are electrophilic, as shown in the drawing on the right. Indeed, the dipolar nature of the O–H bond is such that alcohols are much stronger acids than alkanes (by roughly 1030 times), and nearly that much stronger than ethers (oxygen substituted alkanes that do not have an O–H group). The most reactive site in an alcohol molecule is the hydroxyl group, despite the fact that the O–H bond strength is significantly greater than that of the C–C, C–H and C–O bonds, demonstrating again the difference between thermodynamic and chemical stability.
For a discussion of how acidity is influenced by molecular structure Click Here.
Electrophilic Substitution at Oxygen
Substitution of the Hydroxyl Hydrogen
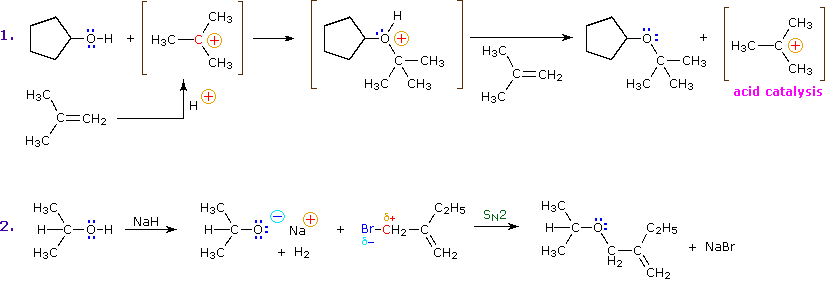
Because of its enhanced acidity, the hydrogen atom on the hydroxyl group is rather easily replaced by other substituents. A simple example is the facile reaction of simple alcohols with sodium (and sodium hydride), as described in the first equation below. Another such substitution reaction is the isotopic exchange that occurs on mixing an alcohol with deuterium oxide (heavy water). This exchange, which is catalyzed by acid or base, is very fast under normal conditions, since it is difficult to avoid traces of such catalysts in most experimental systems.
|
2 R–O–H + 2 Na
|
|
R–O–H + D2O
|
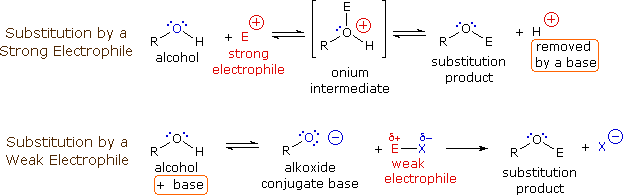
The mechanism by which many substitution reactions of this kind take place is straightforward. The oxygen atom of an alcohol is nucleophilic and is therefore prone to attack by electrophiles. The resulting "onium" intermediate then loses a proton to a base, giving the substitution product. If a strong electrophile is not present, the nucleophilicity of the oxygen may be enhanced by conversion to its conjugate base (an alkoxide). This powerful nucleophile then attacks the weak electrophile. These two variations of the substitution mechanism are illustrated in the following diagram.
The preparation of tert-butyl hypochlorite from tert-butyl alcohol is an example of electrophilic halogenation of oxygen, but this reaction is restricted to 3°-alcohols because 1° and 2°-hypochlorites lose HCl to give aldehydes and ketones. In the following equation the electrophile may be regarded as Cl(+).
(CH3)3C–O–H + Cl2 + NaOH
![]() (CH3)3C–O–Cl + NaCl
+ H2O
(CH3)3C–O–Cl + NaCl
+ H2O
Alkyl substitution of the hydroxyl group leads to ethers. This reaction provides examples of both strong electrophilic substitution (first equation below), and weak electrophilic substitution (second equation). The latter SN2 reaction is known as the Williamson Ether Synthesis, and is generally used only with 1°-alkyl halide reactants because the strong alkoxide base leads to E2 elimination of 2° and 3°-alkyl halides.
One of the most important substitution reactions at oxygen is ester formation resulting from the reaction of alcohols with electrophilic derivatives of carboxylic and sulfonic acids. The following illustration displays the general formulas of these reagents and their ester products, in which the R'–O– group represents the alcohol moiety. The electrophilic atom in the acid chlorides and anhydrides is colored red. Examples of specific esterification reactions may be selected from the menu below the diagram, and will be displayed in the same space.
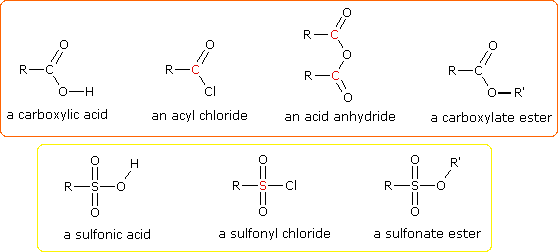
- Examples:
Hydroxyl Group Substitution
Nucleophilic Substitution of the Hydroxyl Group
Using the chemical behavior of alkyl halides as a reference, we are encouraged to look for analogous substitution and elimination reactions of alcohols. The chief difference, of course, is a change in the leaving anion from halide to hydroxide. Since oxygen is slightly more electronegative than chlorine (3.5 vs. 2.8 on the Pauling scale), we expect the C-O bond to be more polar than a C-Cl bond. Furthermore, an independent measure of the electrophilic character of carbon atoms from their nmr chemical shifts (both 13C & alpha protons), indicates that oxygen and chlorine substituents exert a similar electron-withdrawing influence when bonded to sp3 hybridized carbon atoms. Despite this promising background evidence, alcohols do not undergo the same SN2 reactions commonly observed with alkyl halides. For example, the rapid SN2 reaction of 1-bromobutane with sodium cyanide, shown below, has no parallel when 1-butanol is treated with sodium cyanide. In fact ethyl alcohol is often used as a solvent for alkyl halide substitution reactions such as this.
|
CH3CH2CH2CH2–Br + Na(+) CN(–) |
|
CH3CH2CH2CH2–OH + Na(+) CN(–) |
The key factor here is the stability of the leaving anion (bromide vs. hydroxide). We know that
HBr is a much stronger acid than water (by more than 18 powers of ten), and this difference will
be reflected in reactions that generate their conjugate bases. The weaker base, bromide anion, is
more stable and its release in a substitution or elimination reaction will be much more favorable
than that of hydroxide ion, a stronger and less stable base.
Clearly, an obvious step toward improving the reactivity of alcohols in
SN2 reactions would be to modify the –OH functional group in a way that improves its
stability as a leaving anion. One such modification is to conduct the substitution reaction in
strong acid so that –OH is converted to –OH2(+). Since the hydronium ion
(H3O(+)) is a much stronger acid than water, its conjugate base
(H2O) is a better leaving group than hydroxide ion. The only problem with this strategy
is that many nucleophiles, including cyanide, are deactivated by protonation in strong acid,
effectively removing the nucleophilic co-reactant needed for the substitution. The strong acids
HCl, HBr and HI are not subject to this difficulty because their conjugate bases are good
nucleophiles and are even weaker bases than alcohols. The following equations illustrate some
substitution reactions of alcohols that may be effected by these acids. As was true for alkyl
halides, nucleophilic substitution of 1°-alcohols proceeds by an SN2 mechanism, whereas
3°-alcohols react by an SN1 mechanism. Reactions of 2°-alcohols may occur by both
mechanisms and often produce some rearranged products. The numbers in parentheses next to the
mineral acid formulas represent the weight percentage of a concentrated aqueous solution, the form
in which these acids are normally used.
|
CH3CH2CH2CH2–OH + HBr
(48%)
|
|
(CH3)3C–OH + HCl (37%)
|
Although these reactions are sometimes referred to as "acid-catalyzed" this is not strictly correct. In the overall transformation a strong HX acid is converted to water, a very weak acid, so at least a stoichiometric quantity of HX is required for a complete conversion of alcohol to alkyl halide. The necessity of using equivalent quantities of very strong acids in this reaction limits its usefulness to simple alcohols of the kind shown above. Alcohols having acid sensitive groups would, of course, not tolerate such treatment. Nevertheless, the idea of modifying the -OH functional group to improve its stability as a leaving anion can be pursued in other directions. The following diagram shows some modifications that have proven effective. In each case the hydroxyl group is converted to an ester of a strong acid. The first two examples show the sulfonate esters described earlier. The third and fourth examples show the formation of a phosphite ester (X represents remaining bromines or additional alcohol substituents) and a chlorosulfite ester respectively. All of these leaving groups (colored blue) have conjugate acids that are much stronger than water (by 13 to 16 powers of ten) so the leaving anion is correspondingly more stable than hydroxide ion. The mesylate and tosylate compounds are particularly useful in that they may be used in substitution reactions with a wide variety of nucleophiles. The intermediates produced in reactions of alcohols with phosphorus tribromide and thionyl chloride (last two examples) are seldom isolated, and these reactions continue on to alkyl bromide and chloride products.
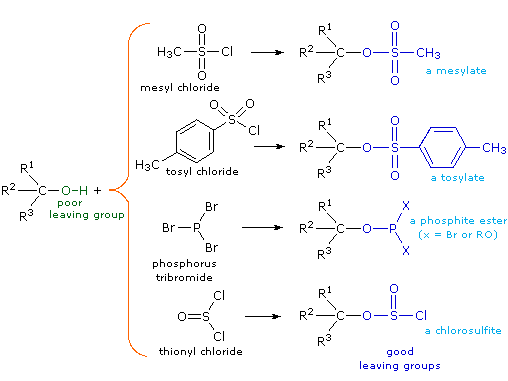
The importance of sulfonate ester intermediates in general nucleophilic substitution reactions of alcohols may be illustrated by the following conversion of 1-butanol to pentanenitrile (butyl cyanide), a reaction that does not occur with the alcohol alone (see above). The phosphorus and thionyl halides, on the other hand, only act to convert alcohols to the corresponding alkyl halides.
CH3CH2CH2CH2–OSO2CH3Na(+)CN(–)
CH3CH2CH2CH2–CN + CH3SO2O(–) Na(+)
Some examples of alcohol substitution reactions using this approach to activating the hydroxyl
group are shown in the following diagram. The first two cases serve to reinforce the fact that
sulfonate ester derivatives of alcohols may replace alkyl halides in a variety of SN2
reactions. The next two cases demonstrate the use of phosphorus tribromide in converting alcohols
to bromides. This reagent may be used without added base (e.g. pyridine), because the phosphorous
acid product is a weaker acid than HBr. Phosphorous tribromide is best used with 1°-alcohols,
since 2°-alcohols often give rearrangement by-products resulting from competing SN1
reactions. Note that the ether oxygen in reaction 4 is not affected by this reagent;
whereas, the alternative synthesis using concentrated HBr cleaves ethers. Phosphorus trichloride
(PCl3) converts alcohols to alkyl chlorides in a similar manner, but thionyl chloride
is usually preferred for this transformation since the inorganic products are gases (SO2
& HCl). Phosphorus triiodide is not stable, but may be generated in situ from a mixture
of red phosphorus and iodine, and acts to convert alcohols to alkyl iodides. The last example
shows the reaction of thionyl chloride with a chiral 2°-alcohol. The presence of an organic base
such as pyridine is important, because it provides a substantial concentration of chloride ion
needed for the final SN2 reaction of the chlorosufite intermediate. In the absence of
base chlorosufites decompose on heating to give the expected alkyl chloride with
retention of configuration
Tertiary alcohols are not commonly used for substitution
reactions of the kind discussed here, because SN1 and E1 reaction paths are dominant
and are difficult to control. This aspect of alcohol chemistry will be touched upon in the next
section.
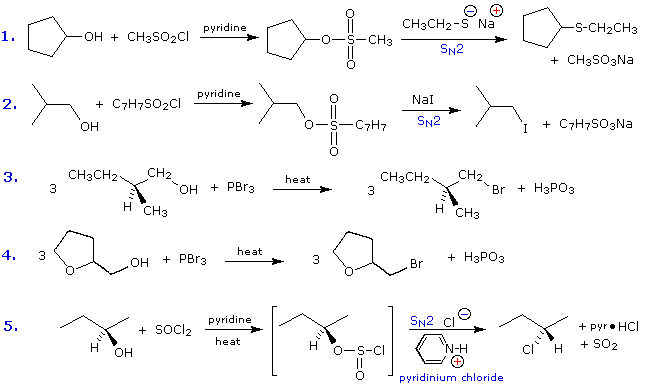
The importance of sulfonate esters as intermediates in many substitution reactions cannot be overstated. A rigorous proof of the configurational inversion that occurs at the substitution site in SN2 reactions makes use of such reactions. An example of such a proof will display above when the An Inversion Proof button beneath the diagram is pressed. Abbreviations for the more commonly used sulfonyl derivatives are given in the following table.
| Sulfonyl Group | CH3SO2– | CH3C6H4SO2– | BrC6H4SO2– | CF3SO2– |
|---|---|---|---|---|
| Name & Abbrev. | Mesyl or Ms | Tosyl or Ts | Brosyl or Bs | Trifyl or Tf |
For a more complete discussion of hydroxyl substitution reactions, and a description of other selective methods for this transformation Click Here.
Elimination Reactions of Alkyl Halides
Elimination Reactions of Alcohols
In the discussion of alkyl halide reactions we noted that 2° and 3°-alkyl halides experienced
rapid E2 elimination when
treated with strong bases, such as hydroxide and alkoxides. Alcohols do not undergo such
base-induced elimination reactions and are, in fact, often used as solvents for such reactions.
This is yet another example of how leaving group stability often influences the rate of a
reaction.
When an alcohol is treated with sodium hydroxide, the following acid-base
equilibrium occurs. Most alcohols are slightly weaker acids than water so the left side is
favored.
R–O–H + Na(+) OH(–)![]() R–O(–) Na(+) + H–OH
R–O(–) Na(+) + H–OH
The elimination of water from an alcohol is called dehydration. Recalling that water is a much better leaving group than hydroxide ion, it is sensible to use acid-catalysis rather than base-catalysis to achieve such reactions. Four examples of this useful technique are shown below. Note that hydrohalic acids (HX) are not normally used as catalysts because their conjugate bases are good nucleophiles and may give substitution products. The conjugate bases of sulfuric and phosphoric acids are not good nucleophiles and do not give substitution under the usual conditions of their use.
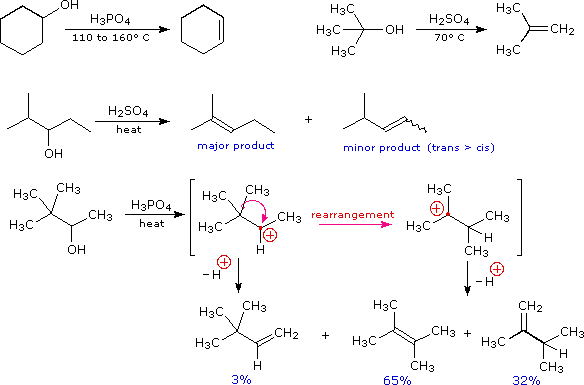
The first two examples (top row) are typical, and the more facile elimination of the 3°-alcohol
suggests predominant
E1 character for the reaction.
This agrees with the tendency of branched 1° and 2°-alcohols to give rearrangement products, as
shown in the last example. The last two reactions also demonstrate that the
Zaitsev Rule applies to alcohol
dehydrations as well as alkyl halide eliminations. Thus the more highly-substituted double bond
isomer is favored among the products.
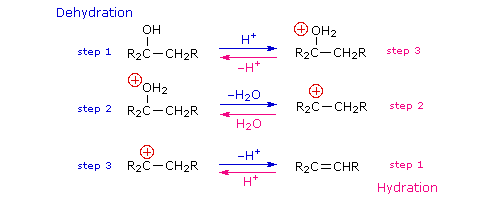
It should be noted that the acid-catalyzed dehydrations discussed here are
the reverse of the
acid-catalyzed hydration reactions of alkenes. Indeed, for reversible reactions such as this the laws of thermodynamics require that the
mechanism in both directions proceed by the same reaction path. This is known as
the principle of microscopic reversibility. To illustrate, the following diagram lists the
three steps in each transformation. The dehydration reaction is shown by the blue arrows; the
hydration reaction by magenta arrows. The intermediates in these reactions are common to both, and
common transition states are involved. This can be seen clearly in the energy diagrams depicted by
clicking the button beneath the equations.
Base induced E2 eliminations of alcohols may be achieved if their sulfonate ester derivatives are used. This has the advantage of avoiding strong acids, which may cause molecular rearrangement and / or double bond migration in some cases. Since 3°-sulfonate derivatives are sometimes unstable, this procedure is best used with 1° and 2°-mesylates or tosylates. Application of this reaction sequence is shown here for 2-butanol. The Zaitsev Rule favors formation of 2-butene (cis + trans) over 1-butene.
CH3CH2CH(CH3)–OSO2CH3C2H5O(–)Na(+)
CH3CH=CHCH3 + CH3CH2CH=CH2 + CH3SO2O(–) Na(+) + C2H5OH
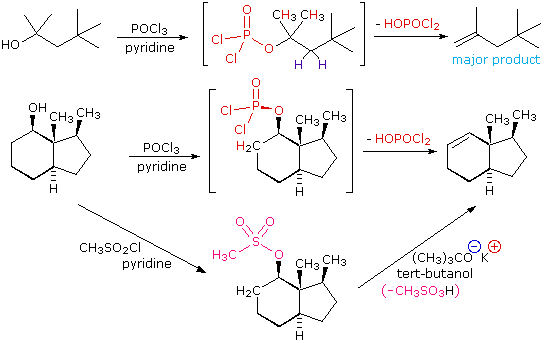
The E2 elimination of 3°-alcohols under relatively non-acidic conditions may be accomplished by treatment with phosphorous oxychloride (POCl3) in pyridine. This procedure is also effective with hindered 2°-alcohols, but for unhindered and 1°-alcohols an SN2 chloride ion substitution of the chlorophosphate intermediate competes with elimination. Some examples of these and related reactions are given in the following figure. The first equation shows the dehydration of a 3°-alcohol. The predominance of the non-Zaitsev product (less substituted double bond) is presumed due to steric hindrance of the methylene group hydrogens, which interferes with the approach of base at that site. The second example shows two elimination procedures applied to the same 2°-alcohol. The first uses the single step POCl3 method, which works well in this case because SN2 substitution is retarded by steric hindrance. The second method is another example in which an intermediate sulfonate ester confers halogen-like reactivity on an alcohol. In every case the anionic leaving group is the conjugate base of a strong acid.
Pyrolytic syn-Eliminations
Ester derivatives of alcohols may undergo unimolecular syn-elimination on heating. To see
examples of these
Click Here