Substituent Effects on the Acidity of Carboxylic Acids
The carboxylic acids are a large and structurally diverse class of compounds. Since most are at least partially soluble in water and have pKa's in the 2 to 5 region, the influence of functional substituents and structural features on aqueous acidity have been studied extensively. Formic acid, HCO2H, is the simplest member of this class, and will serve as a useful reference point, pKa=3.75. Although the greater acidity of formic acid compared with methanol has been attributed to resonance stabilization of the formate anion, the different solvation demands of the respective conjugate anions result in an entropy difference that also favors the formate base. Both factors are depicted in the following illustration.. Resonance delocalization of the negative charge in the formate anion produces a large enthalpic stabilization shown by the magenta arrow. In water solution both methanol and formic acid are incorporated into the dynamic hydrogen bonded structure of liquid water. On ionization, each of these solutes produces a hydrated proton (hydronium ion) and a negatively charged conjugate base. The hydronium ion is common to both cases and can be ignored. The negative charge in the methoxide anion is concentrated on a single oxygen atom and demands strong solvation by water molecules, indicated by the aqua-colored dots. This solvation forces significant structural organization on many water molecules at the cost of decreased entropy. The formate anion also carries a single negative charge, but it is distributed over two oxygen atoms, so the charge density at either site is halved, compared with methoxide. This lower charge density demands much less solvation by water, resulting in a smaller entropy cost.
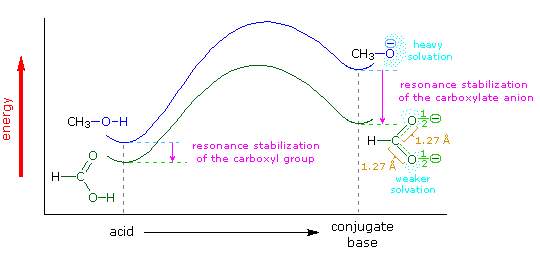
The importance of solvation and the accompanying entropy changes to any discussion of acidity may be seen by comparing the pKa's of methanol and formic acid in water and DMSO, a solvent that poorly solvates anions. In water the pKa of methanol is 15.5, nearly 12 powers of ten less acidic than formic acid (3.75). In DMSO the pKa's of methanol and formic acid are roughly 29 and 13 respectively, representing a very large decrease in Brønsted acid strength for both compounds (more than ten powers of ten). Furthermore, the difference in acid strength between methanol and formic acid in DMSO is magnified about ten thousand times, even though the enthalpic resonance stabilization presumably remains constant. A more extensive discussion of solvent effects on acidity was presented earlier. When comparing the acidities of different acids, care must be taken to use pKa's measured in the same solvent. In this discussion all the pKa's were taken in or extrapolated to water at 25 °C. Measurements in mixed aqueous solvents, using water-soluble organic co-solvents such as ethanol, acetonitrile, dioxane, DMSO and acetone, generally give significantly larger pKa's.
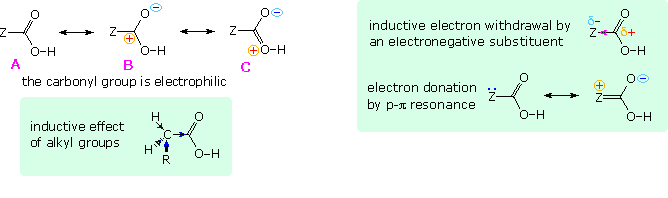
In all other carboxylic acids an organic substituent replaces the hydrogen of formic acid, and it is instructive to analyze the change in acid strength caused by this change. To begin with, we must recognize that the carbonyl moiety of the carboxyl group is electrophilic and withdraws electrons from substituents. The deactivating nature of the carboxyl group on electrophilic substitution of benzoic acid is one example of this property. Resonance structures, such as A, B & C in the following diagram, are often drawn to describe this electrophilic character. The inductive effect of substituent Z in this diagram may enhance or diminish this character, depending on its overall electronegativity. Inductive electron withdrawal will increase the electrophilic character and the acidity of the carboxyl group, as shown in the green shaded box on the right. Resonance electron donation, either by p-π or π-π interaction, would act to stabilize the carboxylic acid, reducing its electrophilicity and acidity. These two effects often act in opposition, and in the case of carbonic acid (H2CO3) electron donation overcomes inductive withdrawal, resulting in a pKa1=6.63.
Saturated aliphatic acids are generally ten times weaker than formic acid, which may seem
surprising since carbon has a higher Pauling electronegativity than hydrogen (2.55 versus 2.20).
However, we must recognize that a carbon atom is larger and more polarizable than hydrogen,
allowing it to shift electrons toward the more electronegative carbonyl carbon of the carboxyl
group. Also, hydrogen and alkyl substituents on the α-carbon assist in this inductive electron
shift, as shown in the green box on the left. This analysis is supported by the activating
influence of alkyl substituents in electrophilic aromatic substitution, the
Markovnikov rule, and the greater
reactivity of aldehydes with nucleophiles compared with equivalent methyl ketones.
The four
carboxylic acids in the first row of the following table illustrate the electron donating quality
of alkyl groups. As the number of carbon atoms in the group increases from one to five, the
inductive electron donation also increases. The compounds in the next three rows of the table
demonstrate that electronegative substituents on an alkyl group can shift its inductive effect
from donating to withdrawing (relative to hydrogen). Thus, all the haloacetic acids are more
acidic than formic acid, with fluoroacetic acid being the most acidic. Additional halogen
substituents have an additive influence, and moving the substituent from the α to a β-carbon
reduces its influence on the acidity. Note that a hydroxyl substituent has a much weaker effect
than any of the halogens, despite the higher electronegativity of oxygen (3.44 compared with 3.16
for chlorine).
| Compound | pKa | Compound | pKa | Compound | pKa | Compound | pKa | |||
|---|---|---|---|---|---|---|---|---|---|---|
| CH3CO2H | 4.76 | CH3CH2CO2H | 4.87 | CH3(CH2)2CO2H | 4.91 | (CH3)3CCO2H | 5.05 | |||
| FCH2CO2H | 2.59 | ClCH2CO2H | 2.85 | BrCH2CO2H | 2.89 | ICH2CO2H | 3.13 | |||
| NCCH2CO2H | 2.50 | HOCH2CO2H | 3.82 | Cl2CHCO2H | 1.25 | Cl3CCO2H | 0.77 | |||
| NCCH2CH2CO2H | 3.98 | ClCH2CH2CO2H | 3.95 | BrCH2CH2CO2H | 4.00 | ICH2CH2CO2H | 4.06 |
Conjugation and Hybridization
The aliphatic acids discussed above do not provide any insight into p-π or π-π conjugation effects, since the sp3-hybridized α-carbon insulates the carboxyl group from such interactions. Conjugation may be studied by using α,β-unsaturated and aromatic carboxylic acids. The two parent compounds of these classes, acrylic acid (CH2=CHCO2H) and benzoic acid (C6H5CO2H), are both slightly stronger than acetic acid and have similar pKa's of 4.26 and 4.20 respectively. Since their influence is probably a combination of inductive and resonance effects, it would be helpful to evaluate one of these alone. The following four compounds represent acetic acid derivatives in which a methyl hydrogen has been replaced with a methyl group, a vinyl group, a phenyl group and a chlorine atom respectively. In each compound a methylene group insulates the substituent from the carboxyl group, prohibiting conjugative interactions. As noted above, the methyl substituent is weakly electron donating and the chlorine exerts a strong electron withdrawing influence. Comparatively, the vinyl and phenyl groups have an electron withdrawing inductive effect roughly 25% that of chlorine. From this we may conclude that resonance electron donation to the carboxyl function in acrylic acid and benzoic acid substantially dilutes the inductive effect of the sp2 substituent groups.
|
The increased electronegativity of sp2 and sp hybridized carbon compared with sp3
carbon was noted earlier. This increase is particularly dramatic for triply
bonded substituents, as seen in the acidity of 2-propynoic acid, HC≡CCO2H, and
3-butynoic acid, HC≡CCH2CO2H, having respective pKa's of 1.90 and
3.30. Conjugative electron donation in 2-propynoic acid is very small, compared with acrylic acid,
reflecting the poor electron donating character of the
triple bond.
Another aspect of
conjugation concerns the ability of a double bond, triple bond or aromatic ring to transmit the
influence of a remote substituent to the carboxyl group. The compounds in the following table
provide information bearing on this issue. The top row consists of β-substituted acrylic acid
derivatives. Methyl and phenyl substituents exert a weakening effect; whereas chlorine strengthens
the acid. Comparing these relationships with similar substituent effects in equivalent saturated
acids (previous table) leads to some interesting differences.
• A
β-chlorine substituent exerts the same acidity strengthening effect regardless of unsaturation in
the connecting chain.
• A β-methyl group decreases the acidity of the unsaturated acid ten
fold over that of the saturated analog.
• A β-phenyl group increases the acidity of the
saturated acid, but decreases that of the unsaturated acid by roughly the same degree.
These
observations may be interpreted in several ways. First, the inductive electron withdrawal by
chlorine through a C–C sigma-bond is about the same as through a pi-bond. Second, The inductive
electron donation by a methyl group occurs to a significant degree by
hyperconjugation
or conjugated hyperconjugation. Finally, the curious inversion of the phenyl influence may be
attributed to an exclusive inductive electron withdrawal down the saturated connecting group,
overpowered by a conjugative donation through the unsaturated chain.
| Compound | pKa | Compound | pKa | Compound | pKa | Compound | pKa | |||
|---|---|---|---|---|---|---|---|---|---|---|
| t-CH3CH=CHCO2H | 4.74 | (CH3)2C=CHCO2H | 5.12 | ClCH=CHCO2H | 3.32 | t-C6H5CH=CHCO2H | 4.50 | |||
| p-CH3C6H4CO2H | 4.36 | p-ClC6H4CO2H | 3.98 | p-CH3OC6H4CO2H | 4.48 | p-O2NC6H4CO2H | 3.42 | |||
| m-CH3C6H4CO2H | 4.27 | m-ClC6H4CO2H | 3.82 | m-CH3OC6H4CO2H | 4.10 | m-O2NC6H4CO2H | 3.47 |
The substituted benzoic acids in the above table exhibit many of the same effects noted for the
acrylic acid derivatives. It must, however, be noted that the meta and para-substituent locations
in these compounds are further removed from the carboxyl group, both in distance and number of
connecting bonds, than in the acrylic acid examples. This will reduce the magnitude of any
inductive effects. The para-location permits conjugative interaction of the substituent with the
carboxyl function; the meta location does not. For comparison purposes remember that benzoic acid
itself has a pKa = 4.2.
A para-methyl substituent appears to have double the
electron donating effect of a meta-methyl group, again suggesting that conjugative
hyperconjugation may be important. The meta-chlorobenzoic acid isomer is significantly more acidic
than the para-isomer, largely because it is closer to the carboxyl function, and in part due to
resonance electron donation by the para-chlorine. The two methoxybenzoic acids are particularly
informative, inasmuch as the meta isomer has a slightly increased acidity, whereas the para-isomer
is significantly weakened. Oxygen has a much larger electronegativity than carbon, but it is an
excellent p-π electron donor to sp2 carbon functions. For the meta isomer, the
inductive effect is somewhat stronger than the resonance donation, but the para-isomer is able to
donate an oxygen electron pair directly into the electrophilic carboxyl function. Both the meta
and para-nitro substituent withdraw electrons from the benzene ring by a combination of inductive
and resonance action, and the corresponding acids are greatly strengthened. A quantitative
treatment of meta and para-substituent effects on the properties and reactions of benzoyl
derivatives has been developed by
L.P. Hammett.
The Ortho Effect
In general, ortho-substituted benzoic acids are stronger acids than their meta and para isomers,
regardless of the nature of the substituent. The ortho effect is large for the nitrobenzoic acids,
which show nearly a 20 fold increase in acidity, roughly an 8 fold factor for the halobenzoic
acids, and a 2.5 to 3 fold increase for methyl and cyano substituents. The methoxybenzoic acids
are exceptional, in that the ortho and meta isomers have nearly identical pKa's (ca.
4.1), presumably due to the exceptional p-π electron donation from oxygen noted above.
Many
of the factors that influence the acidity of substituted benzoic acids are summarized in the
following diagram. First, although the phenyl group is inductively electron withdrawing, it can
donate electrons to a carboxyl group by π-π resonance, as shown in the green shaded box in the
upper left. A substituent Y may perturb the balance of these two factors by its inductive
influence or by resonance. Two resonance cases, one showing electron withdrawal by a nitro
substituent and the other electron donation by a methoxy substituent, are shown to the right of
the green box.
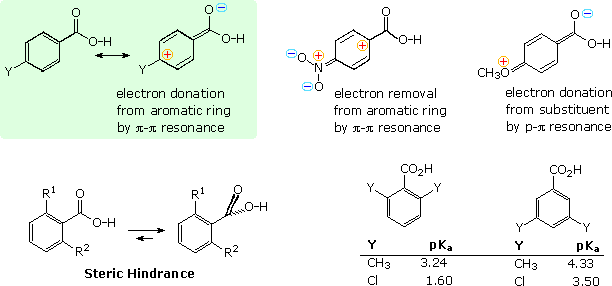
The increased acidity of ortho-substituted benzoic acids is attributed to steric hindrance that
forces the carboxyl group to twist out of the plane of the benzene ring. The inductive character
of the phenyl group does not change with such twisting, but resonance (conjugative electron
donation) requires a coplanar relationship. For example, ortho-toluic acid (R1 =
CH3 & R2 = H) has a pKa of 3.9 compared with 4.2 for benzoic
acid itself. If the methyl is changed to a larger tert-butyl group the pKa drops
to 3. 53. By sandwiching the carboxyl group between two ortho substituents, it is forced to lie
perpendicular to the plane of the aromatic ring, and conjugation is prohibited completely. The
dimethyl and dichlorobenzoic acid isomers shown at the lower right in the diagram provide dramatic
evidence of this conformational effect, with the bis-ortho (2,6-) isomers representing the
exclusive action of the inductive effect.
Steric interference with conjugation may also
perturb the acidity of acyclic unsaturated acids. Thus, 2,3-dimethyl-2-butenoic acid has a pKa
of 4.41, compared with the 5.12 pKa of 3-methyl-2-butenoic acid.
Hydrogen Bonding
The presence of a hydrogen bond donor near a carboxyl group may act to enhance its acidity, as demonstrated by the three isomeric hydroxybenzoic acids and three isomeric benzenedicarboxylic acids (phthalic acids) shown in the following table. Compared with benzoic acid, the meta and para-isomers display expected changes in acidity, due to combined inductive and resonance effects. However, the ortho isomers are both roughly 15 times more acidic, even though the hydroxyl and carboxyl substituents have opposite influences in the para-location.
| ortho relationship | pKa1 | pKa2 | meta relationship | pKa1 | pKa2 | para relationship | pKa1 | pKa2 | ||
|---|---|---|---|---|---|---|---|---|---|---|
| salicylic acid | 2.97 | 13.44 | meta-hydroxybenzoic acid | 4.08 | 9.91 | para-hydroxybenzoic acid | 4.58 | 9.40 | ||
| phthalic acid | 2.98 | 5.28 | isophthalic acid | 3.46 | 4.46 | terephthalic acid | 3.51 | 4.82 |
Intramolecular hydrogen bonding of an ortho OH donor to the carbonyl oxygen of the carboxyl group, acting as an acceptor, increases the positive charge on the carbonyl carbon and consequently the acidity of the carboxyl OH. This is illustrated for salicylic acid in the following diagram. Phthalic acid engages in a similar seven-membered cyclic hydrogen bond with a similar outcome. This intramolecular hydrogen bonding also explains the decreased acidity of the remaining acidic function - that is the phenolic OH in salicylic acid and the second carboxyl group in phthalic acid.
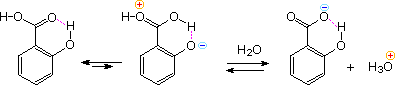
The stereoisomeric 2-butenedioic acids, maleic and fumaric acid display a similar behavior. The cis isomer, maleic acid, has pKa1 = 2.00 and pKa2 = 6.50. This contrasts with the values for the trans-isomer, fumaric acid, pKa1 = 3.00 and pKa2 = 4.50.