Aldol Reactions
The aldol reaction is potentially a powerful synthetic tool. As demonstrated by the following equation, a directed aldol reaction between different aldehydes would produce a single crossed aldol product (the new bond is colored maroon) and create two new stereogenic centers (colored blue). However, conducting this reaction with aqueous NaOH usually generates a mixture of four different adducts, and frequently β-elimination of water when possible.
R1CH2CHO |
+ |
R2CH2CHO |
base |
R1CH2CH(OH)—CH(R2)CHO |
| Carbonyl Acceptor | Enolate Donor | One Crossed Aldol Product |
In this section methods of controlling the enolate donor and carbonyl acceptor reactants to produce a single aldol product will be described and discussed. Several important principles must be taken into consideration for this purpose.
• The initial aldol product may undergo beta-elimination of water, depending on reaction conditions.
• Regioselective enolate anion formation may be achieved by using differences in kinetic vs. equilibrium acidities of α-hydrogens.
• Enolate anions may be trapped as silyl ethers and used subsequently for controlled aldol reactions.
• Enolate-like species may be used for aldol-like reactions with carbonyl compounds, and hydrolyzed later to aldol products.
• Metal cations or other electrophilic moieties may act to orient the reactants in specific ways.
The reversibility of conventional aldol reactions is illustrated by the crossed aldol condensation
of 2-butanone with benzaldehyde, illustrated below. The methyl hydrogens are kinetically more
acidic than the methylene hydrogens, so the α'-enolate is generated more rapidly than the more
substituted α-enolate. As a rule, ketone enolate bases react more rapidly with aldehydes than with
ketones, so self condensation of the ketone is minimal. Both enolate anions add reversibly to
benzaldehyde to form β-hydroxy ketones; however, the α-enolate product (on the left) reverts to
reactants faster than it eliminates water. In contrast, the isomeric α'-enolate product (on the
right) undergoes rapid β-elimination to the observed 5-phenyl-4-penten-3-one final product.
The
significance of aldol reversibility is dramatically demonstrated by the base-catalyzed
isomerization of 2-methyl-1-acetylcyclobutene, a reaction that will be outlined below by clicking
on the diagram.
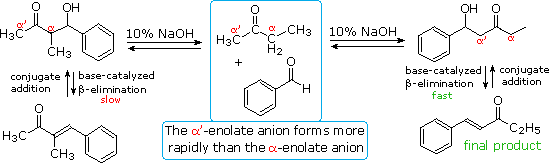
Acid catalysis of the 2-butanone-benzaldehyde reaction gives the isomer, 3-methyl-4-phenyl-3-buten-2-one, as the chief product, thanks to the predominance of the more substituted α-enol as an intermediate. In this case both β-hydroxy ketone intermediates undergo reversible dehydration.
Regioselective Enolate Formation and Diastereoselectivity
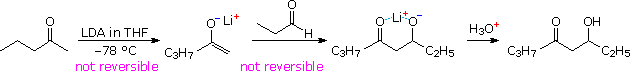
Lithium enolate derivatives of aldehydes or ketones may be formed at low temperature by slow addition of the carbonyl compound to an excess of LDA in THF (-78 °C). In this procedure self aldolization is avoided, because freshly introduced aldehyde (or ketone) reacts with the powerful LDA base more rapidly than with any less basic enolate already present. In the following example, a preformed lithium enolate of 2-pentanone is reacted with propanal at low temperature, ca. -15 °C. Only the desired aldol addition takes place, with very little enolate exchange occurring by proton transfer. Both enolate formation and aldol addition are essentially irreversible, with a forward to reverse rate difference of roughly ten powers of ten (based on pKa values). The lithium salt of the final adduct is quenched in dilute acid to give the β-hydroxyketone. A new stereogenic center is created in this reaction, resulting in a racemic mixture of products.
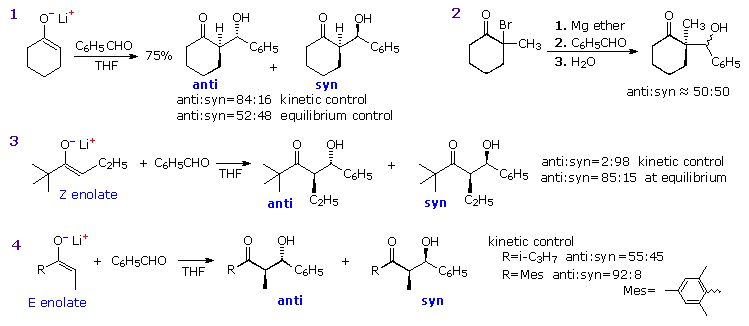
Aldol reactions at an α-methylene group generally create two new stereogenic centers, thus
producing diastereomeric pairs of enantiomers. These diastereomers are called syn and
anti, as described in an
earlier section. The ability to control
the configurational outcome of such reactions is vital to the use of aldol reactions in complex
synthesis, and considerable effort has been directed toward achieving this end. The three
reactions shown in the following diagram illustrate some of the factors to be considered. If the
preformed lithium enolate of cyclohexanone reacts with benzaldehyde under rate controlling
conditions (irreversible addition), the anti isomer is the preferential product (eq. 1). Under
equilibrating conditions, the two diastereomers are produced in nearly equal quantity. Magnesium
and zinc enolates may be prepared by reacting α-bromo carbonyl compounds with these metals, as
illustrated in equation 2. The aldol addition in such cases usually requires a higher temperature.
In all these examples the reactants are achiral and the products are racemic.
Moderately sized cyclic ketones can only generate one enolate stereoisomer
(configuration E), but acyclic ketones may give
E or Z-enolates. In the case of
2,2-dimethyl-3-hexanone, reaction with LDA gives the lithium Z-enolate (98%), as shown in equation
3. Here, the rate controlled aldol reaction strongly favors the syn-diastereomer, but under
equilibrium control the anti-isomer is the dominant product. The size of ketone substituents also
influences the isomer distribution of aldol products, as illustrated by the E-enolate reactions of
the two compounds in equation 4.
From these examples it is clear that kinetic selectivity in the aldol reaction depends in large part on the configuration of the enolate reactant. The first of the following tables illustrates the dramatic influence of the accompanying carbonyl substituent (R) on the Z to E ratio of enolate species formed by reaction of a CH3CH2C=O moiety with LDA. The second table demonstrates how the amide base used for enolate formation may direct reaction exclusively to an E or Z-product, provided the other carbonyl substituent does not exert a controlling influence (e.g. tert-butyl). Identification of enolate species is usually achieved by trapping them as their silyl enol ethers.
| R = | C2H5 | (CH3)2CH | (CH3)3C | C6H5 | CH3O | (CH3)2N |
|---|---|---|---|---|---|---|
| Z:E Ratio | 23:77 | 60:40 | 99:1 | 98:2 | 5:95 | 97:3 |
| Base | LDA | LTMP | LTMP + LiBr | LHMDS | (Et3Si)2NLi | [Ph(CH3)2Si]2NLi |
|---|---|---|---|---|---|---|
| Z:E Ratio | 23:77 | 15:85 | 1:99 | 66:34 | 99:1 | 100:0 |
| LDA = lithium diisopropylamide LTMP = lithium 2,2,6,6-tetramethylpiperidide LHMDS = lithium hexamethyldisilazide | ||||||
An explanation for these interesting and useful characteristics requires consideration of the
transition state for base induced enolate formation. Two putative transition states are displayed
at the top of the following diagram. An early state that resembles reactants is on the left, while
a late state resembling products is on the right. The product from each transition state is the
enolate drawn in the green shaded box between them. Dialkylamide bases such as LDA and LTMP are
powerful bases that react rapidly with simple aldehydes and ketones, presumably by an early
transition state. The chief steric interactions (orange circles) are between R & R1
in the substrate, and between R2 & L (a substituent on the nitrogen base). The
former is particularly strong in tert-butyl ketones, resulting in predominate
Z-enolate formation (the oxygen group is smaller than tert-butyl). For less
sterically demanding R groups, the R2:L interaction acts to favor the
E-enolate, and may dominate when L is large (i.e. LTMP). This tendency is enhanced by added
LiBr.
In contrast, hexaalkyldisilazide bases such as LHMDS,
(Et3Si)2NLi, and [Ph(CH3)2Si]2NLi are much
weaker bases (at least a million times) and will probably react by a later transition state. Here
the Si–N bonds are longer than the C–N bonds of the amide bases, and the entire L2NH
moiety is shifted away from the enolate substrate. Consequently, the L:R2
interaction is relatively unimportant, leading to a preference for Z-enolates.
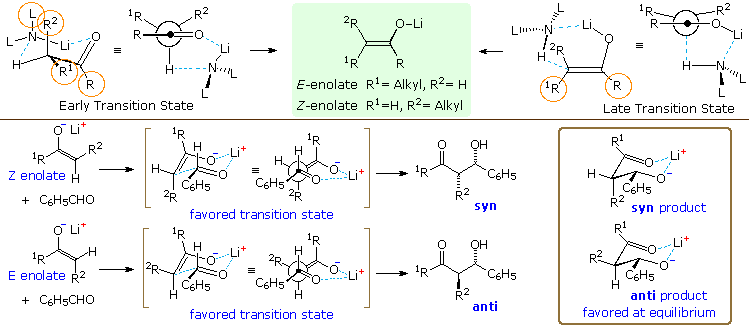
Extensive study of aldol reactions with aldehyde acceptors has led to the general rule: Z-enolates favor syn-products and E-enolates favor anti products. To explain this stereoselectivity a chair-like transition state, called the Zimmerman-Traxler
model, has been proposed, and examples are drawn beneath the enolization transition states in the
above diagram. The lower energy transition state for the Z-enolate aldol has a gauche
relationship between the phenyl and R2 groups, and this leads to the syn-product.
Rotating the aldehyde 180° about the C=O creates an equivalent transition state for the
anti-isomer, which suffers from a destabilizing 1,3-diaxial hinderance between the phenyl and
R1 groups. The favored transition state for an E-enolate aldol has a similar
gauche interaction, but leads to the anti-product. Once the new C–C bond has formed, the aldol
product can relax into a chair-like chelate structure (drawn in the brown box on the right). In
the case of the anti-product, all substituents are equatorial. The equivalent structure for the
syn-isomer has an axial R2 group. In general, anti-aldols are more stable than their
syn analogs. A link to a model for the chair-like transition state is provided
below.
Three additional aldol reactions will be displayed above by
clicking on the diagram. Reaction 5 demonstrates that a very bulky substituent on the α-carbon of
a Z-enolate can dramatically change the stereoselectivity from syn to anti. A similar effect has
been reported for E-enolate reactions. Two modifications of the idealized chair transition
state, the twisted chair and boat models drawn in the box at the upper right, have been suggested
to accommodate these facts. Note that different faces of the carbonyl group bond to the enolate
species in these structures, with the re(si)-face in the twisted boat bonding to the re(si)-face
of the Z-enolate to yield the anti-diastereomer.
Reaction 6 demonstrates
stereoelectronic control in the
bonding of the carbonyl reactant to the enolate intermediate. The bulky t-butyl substituent
maintains an equatorial orientation on the six-membered ring, disclosing axial attack of
benzaldehyde at the α-carbon. Finally, reaction 7 shows an intramolecular
Tischenko reaction following an aldol reaction, the result being
stereoselective construction of a 2-methyl-1,3-diol. The hydride transfer step is rate
determining, so the aldol intermediate is the thermodynamically favored anti-diastereomer. A
six-membered chair-like transition state accounts for the selective reduction of the carbonyl
group.
A cautionary point must be made regarding these models and mechanisms. The lithium cation associated with the amide base and the enolates is organized in oligomeric clusters of substrate and solvent species that are much larger than the atoms (or groups) shown here. These amide and enolate ion clusters have variable compositions, and are presumably in rapid equilibrium with other clusters of the same type.
Silyl Enol Ethers as Enolate Reactants
Enolate anions may be trapped, purified and stored as silyl enol ethers. The most commonly used silyl group is trimethylsilyl (TMS), but other useful derivatives are the tert-butyldimethylsilyl (TBDMS), dimethylphenylsilyl (DMPS) and triisopropylsilyl (TIPS) analogs. As a rule, silyl enol ethers are not as reactive as their anion precursors, but under suitable conditions may be induced to give aldol products when reacted with carbonyl acceptor compounds. The equations above the heavy horizontal line in the following diagram illustrate how mixtures of TMS enol ethers may be prepared under mild conditions, separated by distillation or chromatography, and then used to generate isomerically pure lithium enolates. As expected, the enolates react regioselectively with electrophilic reagents.
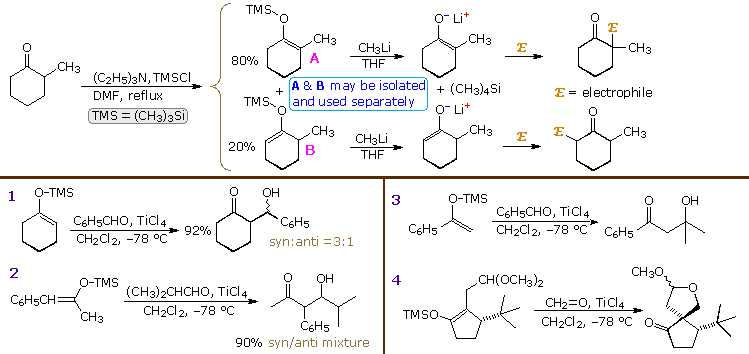
Mixing silyl enol ethers with aldehydes or ketones does not normally result in any reaction, even
at elevated temperatures. In order to effect an aldol-like transformation, it is necessary to
increase the electrophilicity of the carbonyl group by adding a Lewis acid catalyst. Four examples
of such reactions, known as the Mukaiyama aldol, are displayed below the horizontal line.
Reactions 1 and 2 generate a mixture of syn and anti diastereomers. Although there is a preference
for the syn-isomer from both E and Z-enol ethers, this diastereoselectivity is less pronounced
than that of lithium enolates. In contrast to the closed cyclic transition states proposed for the
latter, the acid-catalyzed reactions of the silyl enol ethers are presumed to take place by way of
an open, less organized transition state, which favors syn-products regardless of the enolate
configuration.
Aldehydes are the most common co-reactants with silyl enol ethers, but
catalyzed aldol reactions with ketones have also been reported, as shown in reaction 3. Reaction 4
is an interesting case in which the catalyzed aldol is followed by an acetal exchange involving
the new hydroxyl group. The bulky tert-butyl substituent blocks cis-attack of formaldehyde
on the enolate, directing the new bond formation trans to that group.
Enolborinate Intermediates
The aldol diastereoselectivity of lithium enolates has been attributed to a structurally organized
closed transition state, in which the two oxygen atoms are coordinated to
the metal cation. Since lithium is not an exceptionally strong chelating agent, replacing it with
a larger and stronger electrophile should improve the diastereoselectivity of this important
synthetic method. To this end, enolborinates have proven to be particularly effective. The
comparatively short B–O bond of a dialkyl boron enolate, together with the electrophilic character
of trivalent boron provides a tighter, more highly organized transition state, and in most cases
leads to enhanced diastereoselectivity.
A model of the cyclic aldol transition state may be examined by
Clicking Here
.
The preparation of suitable enolborinates is accomplished by reacting a ketone, ester or
amide with a dialkylchloroborane (or equivalent triflate) and a 3°-amine base, as outlined in the
following equation.. The alkyl substituents on boron are usually n-butyl, cyclopentyl
(Cpen), cyclohexyl (Chex) or the C8H14 substituent of
a 9-borobicyclononane moiety (shown in the diagram). The 3°-amine base is most commonly triethyl
amine (Et3N), N-ethyldiisopropylamine (Hünig's base) or 2,6-dimethylpyridine
(lutidine). Less hindered amines (e.g. pyridine and DABCO) are ineffective, possibly due to
irreversible complexation with the boron reagent.
R1COCH2R2 |
+ |
R2B-Z |
3 °-amine |
R1C(OZ)=CHR2 |
| Ketone | Z=Cl or OTf | E or Z-Enolborinate |
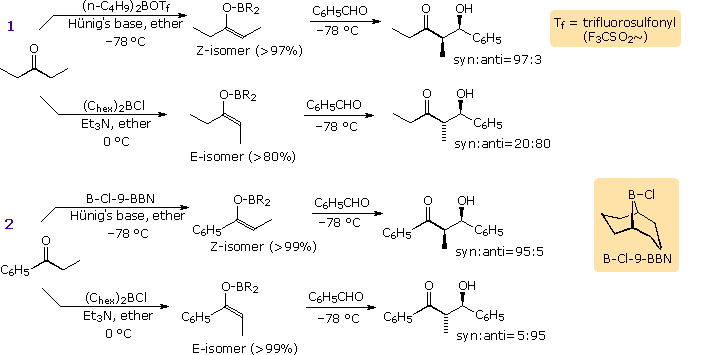
Some examples of the selective formation of enolborinate derivatives from ketones, and their
diastereoselective reaction with aldehydes are displayed in the following diagram. In most cases
the enolborinate is generated in ether solution at –78 °C, and may either be reacted directly with
an aldehyde acceptor or converted to a TMS silyl ether for spectroscopic identification. By
judicious selection of the boron reagent and base, it is clear that specific syn or anti-isomers
may be prepared in high purity and good yield from simple acyclic ketones.
These procedures
also effect regioselective enolborination of unsymmetrical ketones, as demonstrated by the
examples displayed by clicking on the diagram. The dominant enolborinate in all cases is that
derived from the
kinetically favored
enolate anion.
| R | Chex | Chex | Chex | Chex | Chex | 9-BBN | 9-BBN | 9-BBN | 9-BBN | 9-BBN |
|---|---|---|---|---|---|---|---|---|---|---|
| X | OTf | OMs | I | Br | Cl | OTf | OMs | I | Br | Cl |
| Z : E | 25 : 75 | 23 : 77 | 32 : 68 | 11 : 89 | 3 : 97 | 88 : 12 | 82 : 18 | 73 : 27 | 57 : 43 | 56 : 44 |
By clicking on the diagram a second time, a mechanism model for these reactions will appear. The favored chair transition state, T1 or T4, is that in which steric crowding of R1 and R2 is avoided. Large substituents (R) on boron enhance this steric control. Curiously, cyclic ketones which are restricted to E-enolborinate intermediates do not exhibit as large a specificity toward anti-aldol products as do their acyclic counterparts. The cyclohexanone example shown below the horizontal line is typical. The stereospecificity of these aldol reactions is improved by using nonpolar solvents, possibly because the structure of the transition state is tightened due to lack of competitive coordination with boron.
It is not a simple task to write a single mechanism that rationalizes all the experimental results concerning enolborinate formation. Among the variables that must be accommodated are the nature and size of the carbonyl substituents. Thus, tert-butyl ethyl ketones give Z enolates under kinetic control, whereas tert-butyl esters of propanoic acid give E-enolates. Other critical variables are the size and orientation of the alkyl substituents on the R2BX reagent, the nature of the leaving group X, the size of the 3°-amine base, and solvent polarity. A sequence of events that may influence the course of these enolizations is proposed here:
Some Events Associated with Enolborinate Formation
• Acyclic ketones such as 3-pentanone generally adopt a zig-zag conformation. Least motion
enolization would give a Z-enol.
• The nucleophilic oxygen of the ketone coordinates
reversibly with the boron reagent. C=O + R2BX ⇔ C=O–BR2X
• A bulky
boron moiety may shift the conformer equilibrium of the complexed ketone to gauche forms,
favoring an E-enol. [ R=Chex > R=9BBN in size ]
• The B–X bond ionizes to
X(–). This increases the B–O bond strength and the acidity of the α-hydrogens. [
reactivity X=OTf > X=Cl ]
• The 3°-amine base removes an α-proton, forming
the enolborinate. Hünig's base is more hindered and reacts more slowly than triethylamine.
(Note
that the last two events may be concerted, as in an E2 elimination.)
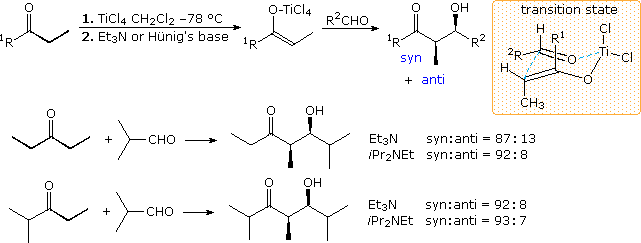
Chlorotitanium Enolates
A convenient procedure for preparing Z-titanium enolate derivatives has been reported by D. A. Evans (Harvard). As shown in the following illustration, these intermediates react with aldehydes to give syn-isomers with high diastereoselectivity. A cyclic transition state similar to that proposed for the enolborinate reactions is suggested.
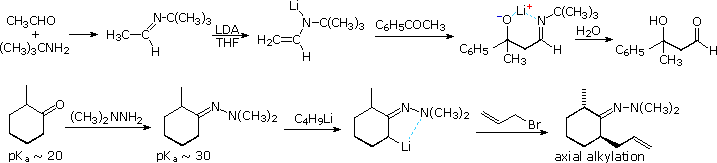
Imine and Enamine Intermediates
Examples of imine and enamine analogs of enolate species are well known. The following diagram gives two examples of metalated imine and hydrazone intermediates in carbon bond forming reactions at an α-carbon. The first is an aldol reaction which would be difficult to accomplish directly. Although the α-hydrogen acidity of aldehydes and ketones do not differ appreciably, aldehydes are much better enolate acceptors than are ketones. By using a preformed tert-butylimine of acetaldehyde as the enolate donor source, the ketone is forced to react as a nucleophile acceptor. Hydrolysis of the imine product generates a disubstituted β-hydroxyaldehyde. In cyclic systems the electrophile bonds preferentially in an axial orientation.
Enamines have been proposed as enolate donors in some aldol reactions. The intramolecular cyclization shown in equation 1 below may be induced by acid or base catalysis or by heating with a 2°-amine such as pyrrolidine. A few drops of acetic acid appear to enhance this catalysis, which probably takes place by the mechanism drawn in the colored box. If the 2°-amine and carboxylic acid functions are incorporated in the same molecule, as for example in the amino acid proline, exceptional catalytic action might be expected. This has been realized in a recent study reported by Alan B. Northrup and David W. C. MacMillan from Cal. Tech. As shown in equation 2, catalytic pyrrolidine effects the homo-condensation of propanal to anti-3-hydroxy-2-methylpentanal under mild conditions. Apparently β-hydroxyaldehydes resist enamine formation, since there is no further reaction of this product. In contrast, alkali metal hydroxides cause polymerization of this aldehyde. Of particular value in this reaction is its' high enantioselectivity. The previously described aldol reactions generate racemic mixtures of stereoisomers from achiral reactants. In this case, enantiomerically pure (S)-proline (the natural amino acid) produces anti-(2S,3S)-3-hydroxy-2-methylpentanal in 99% enantiomeric excess. As expected, (R)-proline catalyzes formation of the enantiomer.
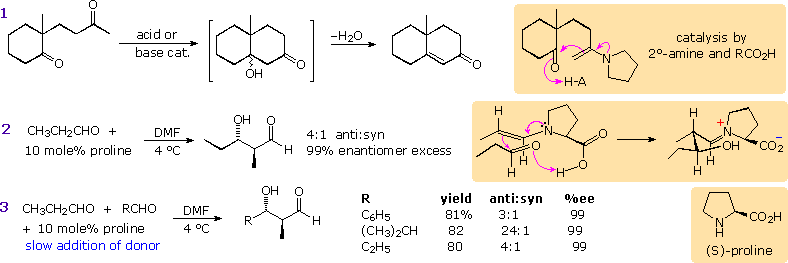
Since the rate of enamine formation from aldehydes is retarded by bulky substituents, cross
condensations with 2-propanal are possible, provided this donor aldehyde is added slowly to the
acceptor-catalyst mixture. Three examples are shown in equation 3. A mechanism for these
stereoselective reactions is drawn in the colored box to the right of equation 2. Proline transfer
from the iminium aldol species to a new aldehyde molecule may be assisted by the small amounts of
water produced in the initial enamine formation.
This transformation is similar to the
aldol-retroaldol processes catalyzed by a family of enzymes called aldolases. By clicking on the
diagram a series of equations illustrating the biosynthesis of 2-deoxyribose-5-phosphate from
glyceraldehyde-3-phosphate by action of the bacterial enzyme DERA will be displayed. Lysine and
aspartic acid functions have been identified in the active site of this enzyme. Configurations of
the reactants and intermediates are not indicated, but these transformations are highly
stereospecific. Antibodies that mimic this enzymatic catalysis have been prepared and used
effectively in enantioselective synthesis.
For information about the influence of chiral reactants on the stereoselectivity of the aldol
reaction;
and the development of enantioselective catalyst systems for this reaction.
Click Here.
The aldol reaction of aldehydes was first reported in 1872 by C.A. Wurtz at the Sorbonne. Over the past quarter century many chemists have contributed to the development of the aldol reaction into the powerful synthetic tool it is now recognized to be. The interested reader should consult the original literature describing their work. Prominent among these are: