Pericyclic Reactions
An important body of chemical reactions, differing from ionic or free radical reactions in a
number of respects, has been recognized and extensively studied. Among the characteristics shared
by these reactions, three in particular set them apart.:
1. They are relatively unaffected by solvent changes, the presence of radical initiators or
scavenging reagents, or (with some exceptions) by electrophilic or nucleophilic catalysts.
2. They proceed by a simultaneous (concerted) series of bond breaking and bond making
events in a single kinetic step, often with high stereospecificity.
3. In agreement with 1 & 2, no ionic, free radical or other discernible
intermediates lie on the reaction path.
Since reactions of this kind often proceed by nearly simultaneous reorganization of bonding electron pairs by way of cyclic transition states, they have been termed pericyclic reactions. The four principle classes of pericyclic reactions are termed: Cycloaddition, Electrocyclic, Sigmatropic, and Ene Reactions. A general illustration of each class will be displayed by clicking on the following diagram. The cycloaddition and ene reactions are shown in their intermolecular format. Corresponding intramolecular reactions, which create an additional ring, are well known.
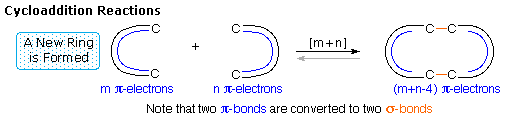
All these reactions are potentially reversible (note the gray arrows). The reverse of a
cycloaddition is called cycloreversion and proceeds by a ring cleavage and conversion of
two sigma-bonds to two pi-bonds. The electrocyclic reaction shown above is a ring forming process.
The reverse electerocyclic ring opening reaction proceeds by converting a sigma-bond to a pi-bond.
As shown, the retro ene reaction cleaves an unsaturated compound into two unsaturated fragments.
Finally, sigmatropic bond shifts may involve a simple migrating group, as shown in the example
above, or may take place between two pi-electron systems (e.g. the Cope rearrangement).
The
general descriptions shown above provide a basis for reaction classification, but care must be
taken to assure that a given transformation is truly concerted. Unfortunately, this is not a
trivial determination, often requiring a combination of isotope labeling and stereochemical
studies to arrive at a plausible conclusion. There is also a subtle distinction to be made between
a synchronous reaction in which all bond-making and bond-breaking events take place in
unison, and a multi-stage concerted process in which some events precede others without
generating an intermediate state.
Although some pericyclic reactions occur spontaneously, most require the introduction of energy in the form of heat or light, with a remarkable product dependence on the source of energy used. An appreciation of the stereoselective structural changes these reactions promote is best achieved by inspecting some individual examples.
Cycloaddition Reactions
 A concerted combination of two π-electron systems to form a ring of atoms having two new σ bonds
and two fewer π bonds is called a cycloaddition reaction. The number of participating π-electrons
in each component is given in brackets preceding the name, and the reorganization of electrons may
be depicted by a cycle of
curved arrows - each representing
the movement of a pair of electrons. These notations are illustrated in the drawing on the right.
The ring-forming cycloaddition reaction is described by blue arrows, whereas the ring-opening
cycloreversion process is designated by red arrows.
Note that the number of curved arrows needed to show the bond reorganization is half the number
total in the brackets.
A concerted combination of two π-electron systems to form a ring of atoms having two new σ bonds
and two fewer π bonds is called a cycloaddition reaction. The number of participating π-electrons
in each component is given in brackets preceding the name, and the reorganization of electrons may
be depicted by a cycle of
curved arrows - each representing
the movement of a pair of electrons. These notations are illustrated in the drawing on the right.
The ring-forming cycloaddition reaction is described by blue arrows, whereas the ring-opening
cycloreversion process is designated by red arrows.
Note that the number of curved arrows needed to show the bond reorganization is half the number
total in the brackets.
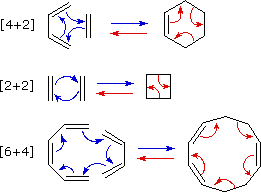
The most common cycloaddition reaction is the [4π+2π] cyclization
known as the Diels-Alder reaction. In
Diels-Alder terminology the two reactants are referred to as the diene and the
dienophile. The following diagram shows two examples of [4π+2π]
cycloaddition, and in the second equation a subsequent light induced [2π+2π]
cycloaddition. In each case the diene reactant is colored blue, and the new σ-bonds in the adduct
are colored red. The stereospecificity of these reactions should be evident. In the first example,
the acetoxy substituents on the diene have identical E-configurations, and they remain cis
to each other in the cyclic adduct. Likewise, the ester substituents on the dienophile have a
trans-configuration which is maintained in the adduct. The reactants in the second equation are
both monocyclic, so the cycloaddition adduct has three rings. The orientation of the quinone
six-membered ring with respect to the bicycloheptane system (colored blue) is
endo, which means it is oriented cis
to the longest or more unsaturated bridge. The alternative configuration is called
exo.
Since the dienophile
(quinone) has two activated double bonds, a second cycloaddition reaction is possible, provided
sufficient diene is supplied. The second cycloaddition is slower than the first, so the monoadduct
shown here is easily prepared in good yield. Although this [4+2] product is stable to further
heating, it undergoes a [2+2] cycloaddition when exposed to sunlight. Note the loss of two
carbon-carbon π-bonds and the formation of two σ-bonds (colored red) in this transformation. Also
note that the pi-subscript is often omitted from the [m+n] notation for the majority of
cycloadditions involving π-electron systems.
![Two [4+2] Diels-Alder examples: diacetoxydiene with fumarate ester, and cyclopentadiene with quinone giving endo adduct then [2+2]](/reusch/virtualtext/img/cycladd1.gif)
By clicking on this diagram two more examples of cycloaddition reactions will be displayed. Reaction 3 is an intramolecular Diels-Alder reaction. Since the diene and dienophile are joined by a chain of atoms, the resulting [4+2] cycloaddition actually forms two new rings, one from the cycloaddition and the other from the linking chain. Once again the addition is stereospecific, ignoring the isopropyl substituent, the ring fusion being cis and endo. The fourth reaction is a [6+4] cycloaddition.
Electrocyclic Reactions
 An electrocyclic reaction is the concerted cyclization of a conjugated π-electron system by
converting one π-bond to a ring forming σ-bond. The reverse reaction may be called electrocyclic
ring opening. Two examples are shown on the right. The electrocyclic ring closure is is designated
by blue arrows, and the ring opening by red arrows. Once again, the number of curved arrows that
describe the bond reorganization is half the total number of electrons involved in the process.
An electrocyclic reaction is the concerted cyclization of a conjugated π-electron system by
converting one π-bond to a ring forming σ-bond. The reverse reaction may be called electrocyclic
ring opening. Two examples are shown on the right. The electrocyclic ring closure is is designated
by blue arrows, and the ring opening by red arrows. Once again, the number of curved arrows that
describe the bond reorganization is half the total number of electrons involved in the process.
In
the first case, trans,cis,trans-2,4,6-octatriene undergoes thermal ring closure to
cis-5,6-dimethyl-1,3-cyclohexadiene. The sterospecificity of this reaction is demonstrated
by closure of the isomeric trans,cis,cis-triene to
trans-5,6-dimethyl-1,3-cyclohexadiene, as noted in the second example.
By clicking on this diagram two examples of thermal electrocyclic opening of cyclobutenes to
conjugated butadienes will be displayed. This mode of reaction is favored by relief of ring
strain, and the reverse ring closure (light blue arrows) is not normally observed. Photochemical
ring closure can be effected, but the stereospecificity is opposite to that of thermal ring
opening.
Sigmatropic Rearrangements
Molecular rearrangements in which a σ-bonded atom or group, flanked by one or more π-electron systems, shifts to a new location with a corresponding reorganization of the π-bonds are called sigmatropic reactions. The total number of σ-bonds and π-bonds remain unchanged. These rearrangements are described by two numbers set in brackets, which refer to the relative distance (in atoms) each end of the σ-bond has moved, as illustrated by the first equation in the diagram below. The most common atom to undergo sigmatropic shifts is hydrogen or one of its isotopes. The second equation in the diagram shows a facile [1,5] hydrogen shift which converts a relatively unstable allene system into a conjugated triene. Note that this rearrangement, which involves the relocation of three pairs of bonding electrons, may be described by three curved arrows.
![Sigmatropic shift notation: [1,5] vs [1,3] hydrogen shifts, plus [1,5] H shift converting an allene to a conjugated triene](/reusch/virtualtext/img/sigtrop1.gif)
By clicking on this diagram two additional examples of thermal [1,5] hydrogen shifts will be
displayed. These reactions are particularly informative in that [1,3] hydrogen shifts are not
observed. The reactant in the first equation is a deuterium labeled 1,3,5-cyclooctatriene. On
heating, this compound equilibrates with its 1,3,6-triene isomer, and the two deuterium atoms are
scrambled among the four locations noted. If [1,3] or [1,7] hydrogen shifts were taking place, the
deuterium atoms would be distributed equally among all eight carbon atoms. On prolonged heating,
or at higher temperatures these cyclooctatrienes undergo electrocyclic ring opening to
1,3,5,7-octatetraene and reclosure to vinyl-1,3-cyclohexadienes.
The second example shows
another [1,5] hydrogen shift, from the proximal methyl group to the carbonyl oxygen atom. The
resulting dienol rapidly exchanges OH for OD before the [1,5] shift reverses. In this manner the
reactive methyl is soon converted to CD3. Since hydrogens alpha to a carbonyl group are
known to undergo
acid or base catalyzed exchange by
way of enol intermediates, we might expect the α'-CH2 group to exchange as well.
However, if care is taken to remove potential acid or base catalysts, the thermal [1,3] shift
necessary for the exchange is found to be very slow.
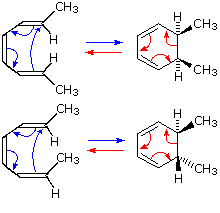
The [3,3] sigmatropic rearrangement of 1,5-dienes or allyl vinyl ethers, known respectively as the
Cope and Claisen rearrangements, are among the most commonly used sigmatropic reactions.
Three examples of the Cope rearrangement are shown in the following diagram. Reactions
1 and 2 (top row) demonstrate the stereospecificity of this reaction. The light blue
σ-bond joins two allyl groups, oriented so their ends are near each other. Since each allyl
segment is the locus of a [1,3] shift, the overall reaction is classified as a
[3,3] rearrangement. The three pink colored curved arrows describe the redistribution of
three bonding electron pairs in the course of this reversible rearrangement. The diene reactant in
the third reaction is drawn in an extended conformation. This molecule must assume a coiled
conformation (as above) before the [3,3] rearrangement can take place. The product of this
rearrangement is an enol which immediately tautomerizes to its keto form. Such variants are termed
the oxy-Cope rearrangement, and are useful because the reverse rearrangement is blocked by
rapid ketonization. If the hydroxyl substituent is converted to an alkoxide salt, the activation
energy of the rearrangement is lowered significantly.
The degenerate or self-replicating Cope
rearrangement has been a fascinating subject of research.
For examples
Click Here
.
![Three Cope [3,3] rearrangement examples, including an oxy-Cope whose enol product tautomerizes to an aldehyde](/reusch/virtualtext/img/sigtrop3.gif)
Two examples of the Claisen Rearrangement may be seen by clicking on the above diagram. Reaction
4. is the classic rearrangement of an allyl phenyl ether to an ortho-allyl phenol. The
methyl substituent on the allyl moiety serves to demonstrate the bonding shift at that site. The
initial cyclohexadienone product immediately tautomerizes to a phenol, regaining the stability of
the aromatic ring. Reaction 5 is an aliphatic analog in which a vinyl group replaces the
aromatic ring. In both cases three pairs of bonding electrons undergo a reorganization.
By clicking on the above diagram a second time two examples of [2,3] sigmatropic rearrangements
will be displayed. The allylic sulfoxide in reaction
6 rearranges reversibly to a less stable sulfenate ester. The weak S-O bond may be
reductively cleaved by trimethyl phosphite to an allylic alcohol and a thiol (not shown). Reaction
7 shows a similar rearrangement of a
sulfur ylide to a cyclic
sulfide. The
[2,3]-Wittig rearrangement is yet
another example.
Ene Reactions
The joining of a double or triple bond to an alkene reactant having a transferable allylic hydrogen is called an ene reaction. The reverse process is called a retro ene reaction. In the bonding direction the ene reaction is characterized by the redistribution of three pairs of bonding electrons. and may be described by a cycle of three curved arrows. As noted earlier, this bond reorganization involves the overall conversion of a π-bond to a σ-bond (or the opposite in the case of retro ene fragmentation). This is the same bond bookkeeping change exhibited by electrocyclic reactions, but no rings are formed or broken in an ene reaction unless it is intramolecular. The following examples illustrate some typical ene reactions, with equation 3 being an intramolecular ene reaction. Ene reactions are favored when the hydrogen accepting reagent, the "enophile", is electrophilic. This is the case for reactions 1 and 2, which proceed under milder conditions than 3, despite the latter's intramolecular nature.
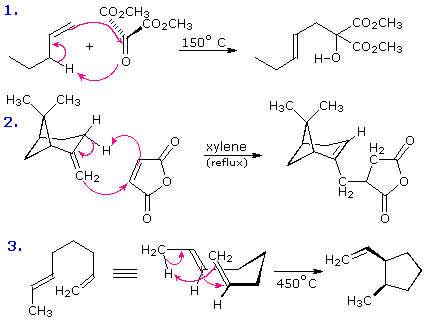
Hydrogen is the most common atom transferred in an ene reaction. Indeed, all the examples shown
above involve hydrogen shifts. Other atoms or groups may, however, participate in ene-like
transformations. Two such cases will be displayed above by clicking on the diagram. Reaction 4 is
drawn as a retro ene reaction, although this has not been demonstrated to be general for all
reactions of allylic alcohols with thionyl chloride. Equation 5 illustrates an unusual "magnesium
ene reaction" in which a Grignard function moves to a new location before reacting with an
electrophilic reagent such as CO2. Because this is an intramolecular ene reaction a new
ring is formed. Clicking on the diagram a second time will display two additional examples.
Equation 6 demonstrates that an enol tautomer, even in low concentration, may function as the
hydrogen donor in the ene reaction. Equation 7 is one of many examples of Lewis acid catalysis in
the ene reaction. A similar acid-catalyzed reaction of simple aldehydes with alkenes to give
allylic alcohols, 1,3-diols or 1,3-dioxanes is known as the
Prins reaction.
Certain retro ene reactions have proven useful as synthetic
transformations. Examples of these concerted elimination reactions may be examined by
Clicking Here
A more elaborate treatment of the intramolecular ene reaction is available by
Clicking Here
Stereochemical Notations
One characteristic shared by most pericyclic reactions, and noted in many cases described above,
is their stereospecificity. This is not the first class of reactions for which a characteristic
stereospecificity has been noted.
Substitution reactions may
proceed randomly or by "inversion" or "retention" of configuration.
Elimination reactions may occur
in an "anti" or "syn" fashion, or may be configurationally random. The terms "syn" and "anti" have
also been applied to
1,2-addition reactions.
Since
these configurational change notations are not appropriate for pericyclic reactions, new
designations are needed. Cycloaddition reactions and sigmatropic rearrangements both
involve pairs of σ-bond-making events (or a coupled bond-making & bond-breaking) associated
with a π-electron system. If all the bonding events take place on the same face of the π-system
the configuration of the reaction is termed suprafacial. If the bonding events occur on
opposite sides or faces of the π-system the reaction is termed antarafacial. Suprafacial
examples of these pericyclic transformations are shown below. The bracketed numbers that designate
reactions of this kind sometimes carry subscripts (s or a) that specify their configuration. Thus
the cycloaddition on the left may be termed a [4s + 2s] process.
Although
cycloaddition reactions are concerted (no intermediate species are formed), the two new bonds are
not necessarily formed in a synchronous fashion. Depending on partial charge distribution in the
diene and dienophile reactants, the formation of one bond may lead the development of the other.
Such unsymmetrical transition state bonding is termed asynchronous.
![Suprafacial [4+2] cycloaddition: diene and dienophile (acrylonitrile) orbital diagram on stacked planes forming the adduct](/reusch/virtualtext/img/supfacl1.gif)
|
![Suprafacial [3,3] sigmatropic Cope rearrangement shown with stacked p-orbital planes and the corresponding skeletal mechanism](/reusch/virtualtext/img/supfacl2.gif)
|
|
| A Suprafacial [4+2] Cycloaddition | A Suprafacial [3,3] Sigmatropic Rearrangement |
|---|
To see a model of a Diels-Alder transition state Click Here .
An example of an antarafacial [1,7] hydrogen shift is shown in the following diagram. The conjugated triene assumes a nearly planar coiled conformation in which a methyl hydrogen is oriented just above the end carbon atom of the last double bond. A [1s,7a] sigmatropic hydrogen shift may then take place, as described by the four curved arrows. With reference to the approximate plane of this π-electron system (defined by the green bonds), the hydrogen atom departs from the bottom face and bonds to the top face, so the transfer is antarafacial.
![Antarafacial [1s,7a] hydrogen shift in a coiled conjugated triene, with orbital diagram showing H moving bottom face to top face](/reusch/virtualtext/img/17antara.gif)
A different notation for configurational change is required for electrocyclic reactions. In these cases a σ-bond between the ends of a conjugated π-electron system is either made or broken with a corresponding loss or gain of a π-bond. For this to happen, the terminal carbon atoms of the conjugated π-electron system must be rehybridized with an accompanying rotation or twisting of roughly 90°. When viewed along the axis of rotation, the two end groups may turn in the same direction, termed conrotatory, or in opposite directions, termed disrotatory. The prefixes con and dis may be remembered by association with their presence in the words concur & disagree. These two modes of electrocyclic reaction are shown in the following diagram in the general form in which they are most commonly observed. Specific examples of these electrocyclic reactions were given earlier.
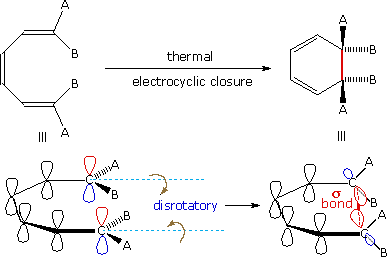
|
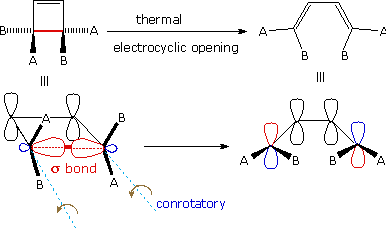
|
|
| A Disrotatory Electrocyclic Closure | A Conrotatory Electrocyclic Opening |
|---|
To see an animation of conrotatory electrocyclic ring closure Click Here .
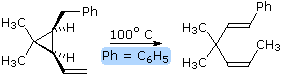 Since ene reactions usually involve coupled bond-making & bond-breaking operations
associated with short π-electron systems (2 or 3 carbons), their stereospecificity is almost
always suprafacial with respect to both components. This configurational feature is illustrated by
the retro ene equation on the right. By clicking on the diagram, a representation of the
transition state for this stereospecific transformation will be drawn. Note that bond-breaking and
bond-making takes place in a suprafacial orientation with respect to each π-electron system. This
reaction is facilitated by the relief of small ring strain.
Since ene reactions usually involve coupled bond-making & bond-breaking operations
associated with short π-electron systems (2 or 3 carbons), their stereospecificity is almost
always suprafacial with respect to both components. This configurational feature is illustrated by
the retro ene equation on the right. By clicking on the diagram, a representation of the
transition state for this stereospecific transformation will be drawn. Note that bond-breaking and
bond-making takes place in a suprafacial orientation with respect to each π-electron system. This
reaction is facilitated by the relief of small ring strain.
Several different structural relationships between the reacting moieties of an intramolecular ene reaction are possible. The examples shown here and above represent the most common orientation. To see examples of two other arrangements Click Here.
Perplexing Features of Pericyclic Reactions
![Three cycloadditions: favorable [8+2], a [6+2] that fails, and an antarafacial [14+2] of a heptafulvalene with tetracyanoethylene](/reusch/virtualtext/img/example1.gif)
The examples of pericyclic reactions presented here provide ample evidence of their usefulness in
constructing or modifying complex molecules, often with a high degree of stereospecificity.
However, in contrast to the general applicability of most common ionic reactions, pericyclic
reactions often display a marked sensitivity to small structural changes. Thus, stereospecificity
may flip-flop, and rates may vary a million fold or more. In the case of cycloaddition reactions,
the three equations on the right illustrate this fact. Equations 1 and 2 show two
very similar transformations, but the first takes place with moderate heat and the second does
not. Note that in each case the triple bond only contributes two electrons to the cycloaddition
transition state. The common [4+2] cycloaddition known as the Diels-Alder reaction proceeds
stereospecifically in a suprafacial fashion, but the [14+2] cycloaddition in
equation 3 is antarafacial with respect to the polyene.
Electrocyclic and sigmatropic
reactions also show puzzling differences in behavior. By clicking on the diagram four examples
will be shown. Equations 4 and 5 describe similar electrocyclic ring openings of
stereoisomeric cyclobutenes. The first occurs under relatively mild heating, but the second
requires extreme heat and may well proceed by bond homolysis to a diradical. Equation
6 shows two electrocyclic ring closures of trans,cis,trans-2,4,6-octatriene. The
thermal reaction is disrotatory, and the photochemical process is conrotatory.
Finally, the
absence of [1,3] sigmatropic shifts of hydrogen was noted earlier, and a
clear example is shown in equation 7. Isomerization of the conjugated triene to toluene
should be strongly exothermic, but a concerted rearrangement of this kind would be a [1,3]
sigmatropic process. In the absence of acid catalysts this triene is completely stable to moderate
heating. Any [1,5] hydrogen shifts that take place reform the starting triene and would require
isotopic labeling to prove. Of course, the isomerization to toluene occurs rapidly if acid is
added.
A Useful Mnemonic Rule
Before pericyclic reactions can be put to use in a predictable and controlled manner, a broad mechanistic understanding of the factors that influence these concerted transformations must be formulated. The simplest, albeit least rigorous, method for predicting the configurational path favored by a proposed pericyclic reaction is based upon a transition state electron count. In most of the earlier examples, pericyclic reactions were described by a cycle of curved arrows, each representing a pair of bonding electrons. The total number of electrons undergoing reorganization is always even, and is either a 4n+2 or 4n number (where n is an integer). Once this electron count is made, the following table may be used for predictions.
|
Thermal |
Transition State Class |
Configurational Preference |
|---|---|---|
| 4n + 2 (aromatic) | Suprafacial or Disrotatory | |
| 4n (antiaromatic) | Antarafacial or Conrotatory | |
|
Photochemical |
Transition State Class |
Configurational Preference |
| 4n + 2 (aromatic) | Antarafacial or Conrotatory | |
| 4n (antiaromatic) | Suprafacial or Disrotatory |
Although this modest mnemonic does not make explicit use of molecular orbitals, more rigorous methods that are founded on the characteristics of such orbitals have provided important insight into these reactions. Since pericyclic reactions proceed by a cyclic reorganization of bonding electron pairs, it is necessary to evaluate changes in the associated molecular orbitals that take place in going from reactants to products. The following section describes approaches of this kind.
Theoretical Models for Pericyclic Reactions
In 1965 R. B. Woodward and Roald Hoffmann of Harvard University proposed and demonstrated that concerted reactions proceed most readily when there is congruence between the orbital symmetries of the reactants and products. In other words, when the bonding character of all occupied molecular orbitals is preserved at all stages of a concerted molecular reorganization, that reaction will most likely take place. The greater the degree of bonding found in the transition state for the reaction, the lower will be its activation energy and the greater will be the reaction rate.
A general introduction to molecular orbitals was presented earlier. The simple compound ethene is made up of six atoms held together by six covalent bonds, as described in the following illustration. A molecular orbital diagram of ethene is created by combining the twelve atomic orbitals associated with four hydrogen atoms and two sp2 hybridized carbons to give twelve molecular orbitals. Six of these molecular orbitals (five sigma & one pi-orbital) are bonding, and are occupied by the twelve available valence shell electrons. The remaining six molecular orbitals are antibonding, and are empty.
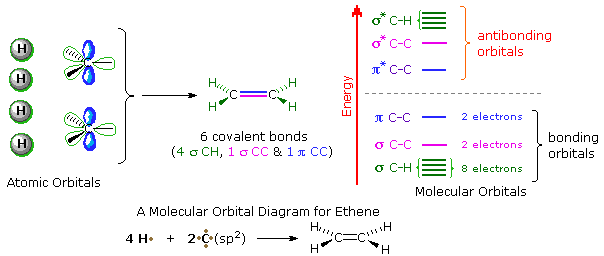
Proper molecular orbitals are influenced by all the nuclei in a molecule, and require consideration of the full structure and symmetry of a molecule for their complete description. For most purposes, this level of treatment is not needed, and more localized orbitals serve well. In the case of ethene and other isolated double bonds, descriptions of the localized π orbitals will be displayed by clicking on the above diagram. Several important characteristics of molecular orbitals need to be pointed out, and this diagram will serve to illustrate them.
1. The spatial distribution of electron density for most occupied molecular orbitals is
discontinuous, with regions of high density separated by regions of zero density, e.g.
a nodal plane. The π-orbital on the left has one nodal plane (colored light blue), and
the π*-orbital on the right has a second nodal plane (colored yellow). As a rule,
higher energy molecular orbitals have a larger number of nodal surfaces or nodes.
2. The wave functions that describe molecular orbitals undergo a change in sign at nodal
surfaces. This phase change is sometimes designated by plus and minus signs associated with
discrete regions of the orbital, but this notation may sometimes be confused for an electric
charge. In the above diagram, regions having one phase sign are colored blue, while those having
an opposite sign are colored red.
3. These localized orbitals may be classified by two independent
symmetry operations ; a mirror plane
perpendicular to the functional plane and bisecting the the molecule (colored yellow above), and
a two-fold axis of rotation (C2) created by the intersection of this mirror plane
with the common nodal plane (colored light blue). The π-orbital on the left is symmetric
(S) with respect to the mirror plane, but antisymmetric (A) when rotated 180°, a
C2 operation. The opposite is true for the π*-orbital on the right, which
has a mirror plane symmetry of A and a C2 symmetry of S. Such symmetry
characteristics play an important role in creating the orbital diagrams used by Woodward and
Hoffmann to rationalize pericyclic reactions.
A model of the p and π orbitals of a double bond may be examined by Clicking Here .
The original approach of Woodward and Hoffmann involved construction of an "orbital correlation diagram" for each type of pericyclic reaction. The symmetries of the appropriate reactant and product orbitals were matched to determine whether the transformation could proceed without a symmetry imposed conversion of bonding reactant orbitals to antibonding product orbitals. If the correlation diagram indicated that the reaction could occur without encountering such a symmetry-imposed barrier, it was termed symmetry allowed. If a symmetry barrier was present, the reaction was designated symmetry-forbidden. Two related methods of analyzing pericyclic reactions are the transition state aromaticity approach, and the frontier molecular orbital approach. Each of these methods has merit, and a more detailed description of each may be examined by clicking the appropriate button below.
Some Examples
Before reviewing representative examples of various types of pericyclic reactions, the previous caution that a given transformation be truly concerted must be emphasized again. The two equations shown in the following diagram describe [2+2] cycloaddition reactions. The second example is particularly interesting because a [4+2] Diels-Alder cycloaddition is possible, but provides only a minor product. A careful examination of these reactions, using probes for ionic and radical intermediates, has shown that these are not concerted transformations. The dipolar and diradical intermediates proposed for these reactions will be illustrated by clicking on the diagram.
![Two non-concerted [2+2] cycloadditions: an enamine with acrylonitrile, and butadiene with tetrafluoroethylene giving a cyclobutane](/reusch/virtualtext/img/stepwis1.gif)
By clicking on the above diagram a second time, an apparent electrocyclic ring opening reaction will be shown. The symmetry favored conrotatory concerted path would generate a very strained trans-cyclohexene double bond, and is energetically unlikely. Instead, a higher activation energy bond cleavage to a diradical intermediate takes place on heating. The racemic diastereomer of this compound undergoes the same ring opening at a lower temperature, and this is believed to be a concerted conrotatory electrocyclic reaction..
With this caveat in mind, extensive lists of pericyclic reactions may be assembled, and their
rationalization by the previously noted mnemonic or orbital analysis is both remarkably successful
and instructive. Many of the reactions cited earlier, together with additional examples, will be
displayed by clicking on the appropriate button.
More about Pericyclic Reactions
An outstanding treatment of pericyclic reactions, including lots of challenging questions with
answers, has been provided by Henry Rzepa.
To reach his Imperial College home page
Click Here.
The link to Dr. Rzepas pericyclic reaction site will be found under
Teaching, Training and Internet Innovation Highlights.
If you have trouble finding
this site, try the menu bar that will be presented by
Clicking Here.