Factors Influencing Carbocation Stability
Carbocation intermediates, sometimes called carbonium ions, have been proposed or identified as important species in reactions ranging from electrophilic addition to alkenes, to unimolecular solvolysis of alkyl halides. The relative stability of these cationic intermediates varies markedly with the presence of substituents on the trivalent charged carbon atom, in a fashion that has led useful empirical rules for predicting reaction selectivity (summarized elsewhere). This stability order for simple alkyl alkenyl and aryl substituted carbocations is repeated below, followed by a table of supportive data.
|
Carbocation Stability |
CH3(+) | < | CH3CH2(+) | < | (CH3)2CH(+) | ≈ | CH2=CH-CH2(+) | < | C6H5CH2(+) | ≈ | (CH3)3C(+) |
|---|
| Cation | Relative Stability |
Tosylate Derivative | Relative Rate of Solvolysis in CF3CO2H |
|---|---|---|---|
| CH3CH2 (+) | 0 kcal/mole | CH3CH2OTs | 1.0 |
| CH3CH2CH2 (+) | 6 | CH3CH2CH2OTs | 5.3 |
| (CH3)2CH (+) | 22 | (CH3)2CHOTs | 2.6 * 104 |
| (CH3)3C (+) | 40 | (CH3)3COTs | 6.3 * 1013 |
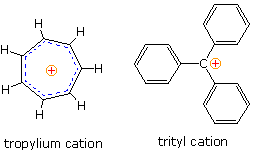 A few carbocations, such as tropylium and trityl (triphenylcarbenium) shown on the right, are
sufficiently stable to form isolable salts with poorly nucleophilic anions, such as
tetrafluoroborate (BF4(–)). However, most carbocations are unstable and
very reactive under normal laboratory conditions, so conventional studies of all but the most
stable of these species have not been possible. Nevertheless, gas phase ionization energies of
alkyl chlorides, hydride affinity measurements (gas phase), molecular orbital calculations, and
low temperature
nmr examination of ionized
alkyl halides in mixed solvents composed of SbF5, SO2, SO2F2
& SO2FCl. (referred to as "super acids") have confirmed the qualitative
relationship shown above. At low temperatures, 1H and 13C nmr spectra of
(CH3)3C(+) and (CH3)2CH(+) were obtained and
interpreted. The charged tricoordinate carbon atom exhibited a 13C signal over 300ppm
downfield from TMS.
A few carbocations, such as tropylium and trityl (triphenylcarbenium) shown on the right, are
sufficiently stable to form isolable salts with poorly nucleophilic anions, such as
tetrafluoroborate (BF4(–)). However, most carbocations are unstable and
very reactive under normal laboratory conditions, so conventional studies of all but the most
stable of these species have not been possible. Nevertheless, gas phase ionization energies of
alkyl chlorides, hydride affinity measurements (gas phase), molecular orbital calculations, and
low temperature
nmr examination of ionized
alkyl halides in mixed solvents composed of SbF5, SO2, SO2F2
& SO2FCl. (referred to as "super acids") have confirmed the qualitative
relationship shown above. At low temperatures, 1H and 13C nmr spectra of
(CH3)3C(+) and (CH3)2CH(+) were obtained and
interpreted. The charged tricoordinate carbon atom exhibited a 13C signal over 300ppm
downfield from TMS.
Inductive & Resonance Effects
What are the factors that influence carbocation stability? The most common means of stabilizing an ion is by charge delocalization, either by inherent structural interactions or by solvation. As noted elsewhere, such structural interactions may usually be classified as inductive or resonance effects, and these may complement or oppose each other. Examples of both are given in the following diagram.
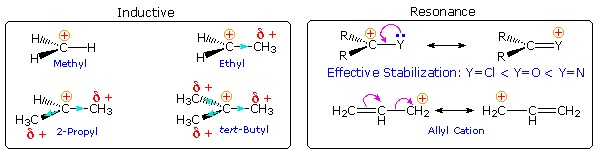
Alkyl groups have somewhat lower electronegativities and are more polarizable than hydrogen. If an
alkyl group is bonded to the carbocation center, the electron pair of the C-C sigma bond will
shift toward the positive charge, transferring a small part of that charge to the alkyl group. In
the diagram on the left above, this inductive electron shift is designated by a light blue arrow
head. Additional alkyl groups provide increased inductive charge dispersion, with each group
assuming a share of the charge. Clearly, this analysis supports the stabilizing influence of alkyl
substituents on carbocations.
Resonance stabilization by non-bonding electron pairs on
adjacent heteroatoms is particularly strong, as shown on the right above. Such charge
delocalization overcomes the potential inductive destabilization of these electronegative
substituents. Similar stabilization is provided by an adjacent nucleophilic pi-electron function,
such as a double bond. A phenyl substituent affords even greater charge delocalization than a
double bond, as reflected by the position of a benzyl cation in the stability order. Resonance
stabilization is generally stronger than inductive effects, and is the predominant factor
stabilizing the tropylium and trityl cations.
Hyperconjugation
Another way in which alkyl substituents stabilize carbocations is illustrated in the following diagram. This conjugative charge delocalization, called hyperconjugation, involves partial pi-bond formation to alpha-carbon atoms, provided suitably oriented C-H or C-C bonds are present. The small increase in stability of the 1-propyl cation compared with an ethyl cation, as noted above, suggests that C-C hyperconjugation provides slightly greater stabilization than does the C-H hyperconjugation shown here. Hyperconjugation by alkyl substituents also acts to stabilize unsaturated functional groups, as noted earlier for carbon-carbon double bonds.
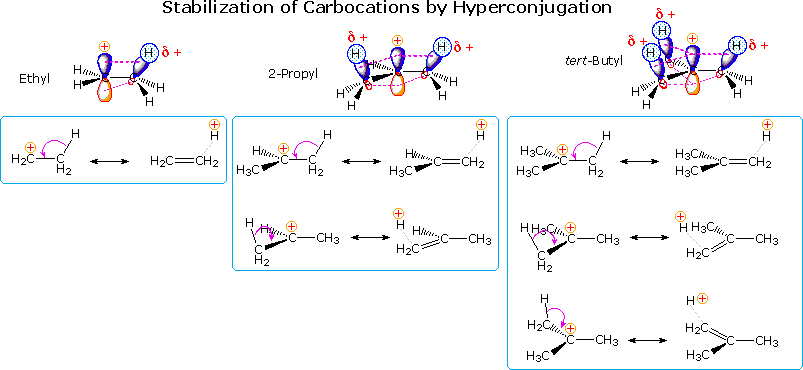
Since hyperconjugation and the inductive effect act in the same manner, their relative importance
in carbocation stabilization is a matter of interest. Some insight to this question is found in a
group of novel compounds, bridgehead substituted bicyclic halides. Two examples of these compounds
are shown below. The nomenclature of bridged bicyclic compounds identifies the length of the
chains that connect the bridgehead atoms (colored pink here). Three connecting chains are present
in a bicyclic compound, and the number of atoms in each chain (excluding hydrogen) is given as a
number in brackets. If the chains are of different lengths the longest is listed first. One of the
bridgehead atoms is numbered one, and the longest chain continues the numbering sequence until the
second bridgehead atom is included. Numbering then continues along the next longest chain. The
base name of the bicyclic compound reflects the total number of carbons, and is therefore the sum
of the bridging chains plus two (the bridgehead atoms).
![Bicyclic nomenclature: bicyclo[m.n.o]alkane bridgehead labeling, 1-chlorobicyclo[2.2.2]octane, 1-bromobicyclo[2.2.1]heptane](/reusch/virtualtext/img/bicyclic1.gif)
The bridgehead substituted halides shown above will form 3°-carbocations when ionized. Inductive
stabilization of these cations should be similar to that of the tert-butyl cation, so if this were
the predominant stabilizing factor from alkyl substitution, the reactivity of these halides should
be similar to their tert-butyl counterparts. In practice, however, the bridgehead halides were
found to be much less reactive. Indeed, 1-chlorobicyclo[2.2.1]heptane was recovered unchanged from
prolonged treatment with hot
ethanolic silver nitrate. The
instability of such bridgehead carbocations has been attributed to the pyramidal shape forced upon
the trigonal carbon (sp2 hybridized). However, covalent bonds are generally able to
accommodate modest bending distortions without significant destabilization, and the inductive
shift of electron pairs toward the positively charged carbon atom is unlikely to be impeded by a
pyramidal configuration of the carbocation.
An alternative explanation is that
hyperconjugation with the alpha-methylene groups is prohibited by the rigid configuration of these
bridgehead cations. By clicking on the diagram above, an illustration of one possible C-H
hyperconjugative interaction will be displayed. The carbon-carbon double bond implicit to this
occurrence is badly twisted, and could not exist as such in any stable alkene. Structural
prohibitions of this kind are encompassed in an empirical guideline called
Bredt's rule.
On the other
hand, C-C hyperconjugation may act to partially stabilize bridgehead carbocations. By clicking on
the diagram a second time, two examples of such hyperconjugation will appear. While still
relatively inert, the bicyclo[2.2.2]octane compound is roughly a million times more reactive than
its bicyclo[2.2.1]heptane analog, shown above. This may be attributed to improved
hyperconjugation, since the appropriate C-C bonds (colored red) are better aligned with a
developing bridgehead carbocation. Furthermore, confirmation of expected changes in bond lengths
resulting from such hyperconjugation has been obtained by X-ray diffraction analysis of a
crystalline SbF6 salt of the adamantane cation. In this study, the red-colored bonds
were lengthened and the green-colored bonds were shortened.